

There are lots of Cleaning Memos stored somewhere in my brain that don’t come to light until I get relevant questions on the subject. The issue of “residues of multiple actives” is one of those things that I have considered in the past, but with all the other problems in cleaning validation, it has never risen to near the top of the pile. However, it now has.
What I mean by “residues of multiple actives” deals with setting limits. Limits for conventional actives (those actives that are not “highly hazardous”) have a limit generally set at 0.001 of the minimum dose of the active in a maximum dose of the next drug product. The conundrum is this: if 0.001 of a dose of a given active is allowed in a drug product, can I allow 0.001 of a dose of Active A and 0.001 of a dose of Active B in the same product? Or taken even further, can I allow 0.001 of a dose of Active A, 0.001 of a dose of Active B, 0.001 of a dose of Active C, 0.001 of a dose of Active D, and so on?
You might ask whether that is a realistic concern. And, as the conventional consulting response goes, “It depends.” One situation where this comes up is where I make a finished drug product with API-D. In the manufacture of that finished drug product, there may be other drug products manufactured before on the same equipment. Therefore, I consider independently the residues of actives in the drug products made immediately before the drug product with API-D. However, because I consider each prior product independently, this does not present a problem. I allow 0.001 of a dose of either API-A, API-B, or API-C from the previous drug products.
Where the conundrum pops up is in API manufacturing when I consider the manufacture of API-D itself. If this is made in non-dedicated equipment, then API-A may contain as much as 0.001 of a dose of another active. Let’s assume that other actives might be API-Q, API-R, or API-S, each of which could be made before the manufacture of API-D. Therefore, when I manufacture a drug product with API-D, it might contain as much residue from the cleaning processes as (for example) 0.001 of a dose of API-Q (from the API manufacturing/ cleaning) and 0.001 of a dose of API-B (from the drug product manufacturing/cleaning). Is this acceptable?
Another situation where this may arise involves just drug product manufacture (for this, I will ignore the impact of prior API manufacture, although it further complicates the issue). Let’s say I am making three drug products, each with a different active: DP-X with API-X, DP-Y with API-Y, and DP-Z with API-Z. Let’s assume (using a simple example) that the products are made in Equipment items #1, #2, and #3 according to the following matrix of what equipment each drug product contacts:
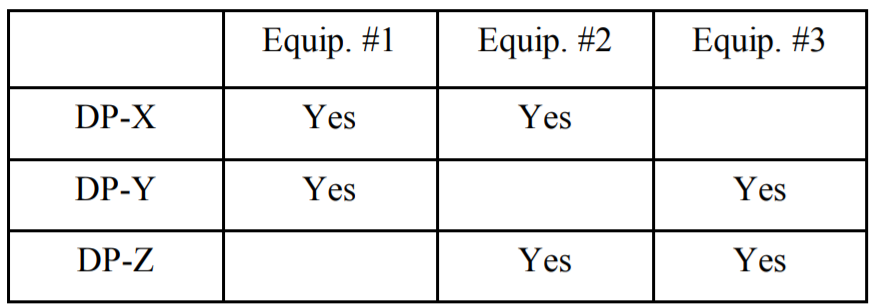
If I limit my carryover calculations only to those equipment items actually shared between any two products, what happens? Let’s just take a look at what residues are allowed in DP-X. From Equipment #1 I will allow 0.001 of a dose of API-Y, and from Equipment #2, I will allow 0.001 of a dose of API-Z. The question is whether this is acceptable.
Before I answer that question, there is one way to possibly get around that question in this latter example. That would involve using the total equipment surface area for the manufacture of DP-X when I calculated the limit. Assuming that Equipment #1 and #2 were equal in surface area, then my calculations would yield limits based on 0.0005 of a dose of API-Y and 0.0005 of a dose of API-Z (meaning a combined amount equal to 0.001 of a “dose”). If they were unequal in surface area, it would not change the basic conclusion. For example, if Equipment #1 had twice the surface area of Equipment #2, then a calculation based on the total equipment surface area for the manufacture of DP-X would yield limits based on 0.00067 of a dose of API-Y and 0.00033 of a dose of API-Z (again meaning a combined amount equivalent to 0.001 of a “dose”).
Let’s get back to the question asked about whether residue levels of 0.001 of a dose of API-Y and 0.001 of a dose of API-Z should be allowed in DP-Z. Let me modify that question slightly to ask whether setting limits of 0.001 of a dose of API-Y and 0.001 of a dose of API-Z should be allowed in DP-Z. My general answer would be that it should be acceptable, provided that there are no unusual drug interactions between API-Y and APIZ (or between API-Y, API-Z, and API-X). My first reason for this opinion is that while the limit of each residue is 0.001 of a dose, the actual level is much lower because most companies achieve, or try to achieve, residue levels less than 25% of the established limit. Furthermore, there are so many “worst-cases” built into cleaning validation programs that it is not likely that actual residue levels will come near 0.001 of a dose.
My second reason is that most programs are designed to achieve low residue levels on the worst-case swab sampling locations. Based on the general principles of stratified sampling (see my March, April, and May 2010 Cleaning Memos), total carryover (assuming uniform contamination of the next product) is much lower than the carryover based on the worst-case (highest residue level) swab sample. This provides an extra margin of safety.
A third consideration is whether limits were established on the medically safe level using a dose carryover criterion, or whether limits were set using a default limit (such as 10 ppm in the next product), even lower than the “medically safe” limit based on 0.001 of the dose. In the case of default limits, there is some level of additional safety built in.
Now it is possible to carry this rationale too far. What if the manufacturing situation were such that the carryover involved 10 different actives, each being allowed at 0.001 of a dose? I would probably draw the line here and not allow that situation. In that case, I would make sure that my calculated limits were based on the total equipment train for the cleaned product, thus limiting the total combined amount to an equivalent of 0.001 of a “dose”.
To put this in perspective, realize that even in the case where I am manufacturing only two drug products on the same equipment using a detergent for cleaning, I am allowing a medically safe level of the other drug active, and I am allowing a medically safe level of the detergent. Is this not similar to allowing a medically safe level of one active and a medically safe level of a different active? And, we can also throw in the possibility of a different active additionally being present in the manufactured API. So this is a situation that is not really so foreign.
However, I believe that based on a risk analysis, most companies would not find this concern to be a significant risk requiring immediate attention with preventive actions required. However, it is one of those things that might require some thought and documentation to show that the concern is not significant.


