

As you should be aware, the ISPE Risk-MaPP dismisses conventional ways of setting limits based on calculations such as 0.001 of a daily dose. It advocates setting limits for both highly hazardous actives (HHAs) and non-highly hazardous actives (NHHAs) using an ADE (Acceptable Daily Exposure). Certainly, the principles of the ADE approach are appropriate for HHAs. Are they applicable to NHHAs?
Let’s review what has been done for HHAs. In the ADE approach, the critical effect is determined. The critical effect for an HHA is usually that property that makes the active highly hazardous. For example, for a drug with reproductive hazards, the critical effect is usually the toxic reproductive property. Then, based on an animal study (or other more relevant human studies), the highest level at which that expression of the critical effect is not seen is the NOAEL (No Observable Adverse Effect Level). Note that this is not a NOEL (No Observable Effect Level), but rather is focused on the adverse effect evaluated in the test procedure (in the example I’ve given, the adverse effect would be some observation of reproductive toxicity).
Then, a variety of “uncertainty factors” are applied based on the judgment of the toxicologist (these are designated UFc, or composite uncertainty factor, in Risk-MaPP). Uncertainty factors account for such issues as interspecies differences, interspecies differences, subchronic to chronic extrapolation, LOAEL to NOAEL extrapolation, and database completeness. In addition to the composite uncertainty factors, a modifying factor (designated MF in Risk-MaPP) is a factor that Risk-MaPP states “may also be considered if there is a need to address residual uncertainties not covered by the other factors”. A third adjustment is a pharmacokinetic factor (designated PK in Risk-MaPP), which can account for different routes of exposure (for example, an injectable drug active for which the animal test is via oral administration). There is also a statement following a discussion of those three factors that “Additional factor(s) may be needed to adjust health-based limits such as ADEs because of the potential for bioaccumulation with repeated exposure.”
Once the ADE is determined, limits in the next product, limits per surface area, etc., are calculated using conventional industry methods. Okay, how is this to be applied to an NHHA? Exactly what is the critical effect for a conventional drug active that is not highly hazardous? Risk-MaPP only gives a hint for this determination. There are four cases in RiskMaPP where an ADE is given for actives that are not highly hazardous. Here are those four cases, with the drug active description, the ADE, and the daily dose of the active (you’ll see why I’m including the daily dose in just a minute). This data is found on page 102 (Section 14.1) of Risk-MaPP.
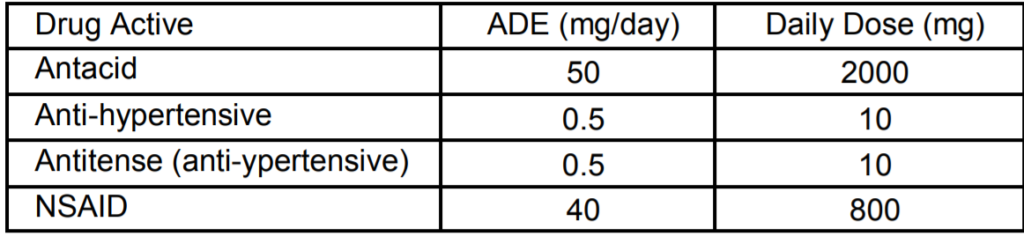
The statement is made that these ADE values were determined by a qualified toxicologist. On page 104, it is further stated that
“A review of all available data on each API (e.g., the data included in the Clinical Investigators Brochure or New Drug Application or the scientific literature) was completed to identify all possible adverse effects that may be produced by the compound. For each compound, the critical effect, i.e., the first adverse effect observed at the low end of the dose-response curve, was identified. The ADE was then derived by dividing the no-effect level for the critical endpoint by appropriate uncertainty or safety factors. Additional adjustments to the ADE were needed depending on the subpopulation it will be used to protect.”
Certainly sounds like the typical Risk-MaPP approach. But is that the whole story? In the next paragraph is a statement about determining an ADE for the new anti-hypertensive:
“Briefly, the ADE was based on the low clinical dose of 10 mg and incorporates appropriate uncertainty factors to address interindividual variability and extrapolation to a no-effect level.”
If I understand this correctly, the ADE in these cases used the lowest clinical dose (which appears to be the daily dose) and applied the various modifying factors to that value. It is unclear how this fits into the way Risk-MaPP proposes for setting ADE values. In my opinion, the daily dose should not be considered a NOAEL value, unless the Risk-MaPP authors are not considering the therapeutic effect of the drug as an “adverse effect” (perhaps on the justification that a for a drug product that lowers blood pressure, the lowering of blood pressure is not an adverse effect). If that is the justification, I find that hard to accept.
In an email exchange with one of the toxicologists for the Risk-MaPP document, I was told (relating to the “new anti-hypertensive” information) that:
“The examples used in the appendices were hypothetical. The ADE of 0.5 mg/day for the antihypertensive with a 10 mg/day therapeutic dose reflects a composite uncertainty factor of 20. Using the approach described in Chapter 5, this could be the result of a chemical-specific adjustment factor of 7 (based on pharmacokinetic and pharmacodynamic data) and a factor of 3 to extrapolate from a LOAEL to a NOAEL. The 10 mg dose would be expected to cause clinically significant cardiovascular effects as well as off-target adverse side effects and would be considered a LOAEL.”
For me, this raised even more red flags. First, that a clinical dose would be considered a LOAEL. I would assume for most marketed products that there would be some evidence of a therapeutic effect at a dose lower than the daily dose. If that is the case, then that lower dose (not the daily dose) would be the LOAEL (again assuming that the therapeutic effect would be considered part of the adverse effect). Second, what does “hypothetical” mean? Does that mean these were not based on real data from a real product? Particularly since Risk-MaPP used this type of data to say that the conventional 0.001 of a dose calculation was inappropriate for cleaning validation purposes, it would seem appropriate (at least to me) that the data presented was not “hypothetical”, but was based on real situations. In other words, it probably is not the case that ADE values for NHHAs are going to be in the range of 0.05 (1/20) to 0.025 (1/40) of a daily dose (as suggested by the examples in Risk-MaPP).
Which get me back to two concerns about ADE values? How are they determined? And, is there too much “judgment” involved in the way they are determined? Or, in the case of calling a daily dose the LOAEL, is inappropriate judgment involved? This concern about the Risk-MaPP allowing too much leeway based on a toxicologist’s judgment was raised (as best I can tell) in the recent EMA document (“Concept Paper on the development of toxicological guidance for use in risk identification in the manufacture of different medicinal products in shared facilities”, 20 October 2011). That document states that the EMA is seeking an approach for setting exposure limits for multiproduct equipment/facilities that “should be scientifically based and aim to limit variability in deriving acceptable exposure limits thereby ensuring consistency.” emphasis added At least one interpretation of this statement is that EMA wants to get away from situation where adjustments factors for ADE values are strictly based on the judgment of the toxicologist, and that more careful delineation of rules should be established so that one toxicologist’s judgment is not significantly different from another toxicologist’s judgment. I would also add that the method for determining the NOAEL for non-highly hazardous drug actives also requires some attention.
On a practical note, my opinion on ADE values is changing. ADE values for NHHAs can be used if they are properly determined (and the examples in Risk-MaPP do not appear to be properly determined). Certainly, they can be employed because they will result in higher limits. But the use of an ADE is not a requirement. The ADE approach certainly allows more flexibility than a one-size-fits-all approach based on 0.001 of a dose being a safe amount. However, I still believe it is the case for non-highly hazardous actives that 0.001 of a dose is a valid approach. It is just more conservative than an individual toxicological evaluation for a given active. That is certainly expected, because we are all aware that the dose/response curve for different actives will have different slopes (and the slope will be important in going from a clinical dose to a NOAEL value).
I imagine the pushback I will hear from ADE advocates is that one doesn’t know that 0.001 of a dose is a safe level unless one determines the ADE. My reply is that, based on the “hypothetical” determinations in RiskMaPP, 0.001 of a dose is a more conservative value. There is a very simple way to resolve this. I suggest a consortium of industry professionals select about 20 common drug actives that are not considered highly hazardous. For those 20 drug actives, select several toxicologists and provide them with relevant toxicity information on those 20 drug actives. Have the toxicologists (independently of each other) determine the ADE value. Those ADE values can then be compared to the 0.001 dose criterion to see whether 0.001 of a dose is appropriately protective. Such a study would also allow the industry to determine how much leeway there is in ADE determination between different qualified toxicologists. This is a challenge I make to the Risk-MaPP advocates and the industry. After all, valid data trumps theory (and speculation) every time.
To put this is perspective (and so you don’t think I’m getting soft in my old age), while the ADE approach is a valid one for cleaning validation limits for NHHAs, it needs more refinement and definition. The Risk-MaPP authors did a very good thing by pointing out that dedication or non-detectability may not be needed for HHAs. They only confused the issue when they tried to advocate (and illustrate) the use of ADEs for NHHAs.


